
Methods

DNA Isolation
To test for the specific VMAT-2 gene a DNA sample must be obtained. The goal of the first procedure was to isolate DNA from cheek cells. DNA Extraction: Salt solution mouth rinse used to obtain cheek cells. (top left photo) Solution centrifuged to collect cells. Chelex solution added to centrifuged pellet. Chelex increases the efficacy of PCR (figure 1) by binding to metal ion cofactors that aid enzymes in the degradation of DNA. The Chelex inhibits hydrolysis of DNA. The solution with the Chelex is then heated to allow the Chelex to bind to dissolved metals within the solution that would otherwise inhibit the PCR reaction and hinder the ability to collect proper data for the overall experiment. Heating the cells denatures them and makes them burst, releasing their DNA into solution. The tube is cooled and centrifuged once more. The liquid at the top of the sample after centrifuging is the DNA. (Bottom photo)
PCR Amplification of VMAT2 Gene
Primer was added to the beaded Taq DNA polymerase and buffers. Once the bead was dissolved, DNA was added. The tube was centrifuged, and then added to the PCR machine. The PCR machine cycles through hot and cooler temperatures to denature DNA, enabling primers to bind to DNA, allowing Taq polymerase to add nucleotides, and repeat. This replicated the desired sections of DNA without replicating the entire strand.
Gel Electrophoresis
Dye buffer was added to the PCR tubes to serve as a visual cue for when electrophoresis was to be terminated (the dye moves faster than the DNA so DNA does not run off the gel). A ladder was added into the first well to serve as a template. The ladder has known lengths that the DNA can be compared to. DNA with dye was added into wells 3 through 6.(top left photo) The gel was then run for approximately 30 minutes. After running, the gel was placed under an ultraviolet light and photographed.(bottom right photo) The photograph was used to determine specific lengths of each DNA piece. Samples were cut out of the gel with a fresh razorblade, inserted into a labeled tube, and refrigerated.
Sample Purification
Gel sample purification was done to obtain a clean DNA sample ready for analysis. QG buffer was used to dissolve the non DNA components of the agarose sample. The sample was heated to completely dissolve any leftover agarose not dissolved by QG. Isopropanol was added to the solution to help the DNA bind. The solution was transferred to a 'column' tube to filter the sample. The column was centrifuged to bring the DNA sample onto the white material in the column. The sample is then washed to remove any other trace elements present in the DNA. PE buffer was added to clean the sample of any extra salts that may have been present and would have interfered with sequencing. The last buffer, EB, was used to release the DNA sample from the white column into a clean eppendorf tube for DNA sequencing. The sample was sent to a lab for DNA sequencing.
Statistical Methods
A Chi-square test was used to compare the student class averages to the averaged data from Hamer’s study. This calculation tests the null hypothesis, proving statistical significance between expected and observed results. Our p-value was greater than .05, the cutoff for statistical significance. This means that our data were not significant. There were six categories tested (one for each of the two possible alleles at each of the three pairs of interest). The degrees of freedom were 5 (n-1). Hamer’s data was obtained from http://pharmacogenetics.ucsf.edu/public-results/vmat2/VMAT2-tab.html and the class snp data was simply a total of allele occurrences. Each of these data sets were converted into percentages per base pair and input into the chi-square calculation.
To test for the specific VMAT-2 gene a DNA sample must be obtained. The goal of the first procedure was to isolate DNA from cheek cells. DNA Extraction: Salt solution mouth rinse used to obtain cheek cells. (top left photo) Solution centrifuged to collect cells. Chelex solution added to centrifuged pellet. Chelex increases the efficacy of PCR (figure 1) by binding to metal ion cofactors that aid enzymes in the degradation of DNA. The Chelex inhibits hydrolysis of DNA. The solution with the Chelex is then heated to allow the Chelex to bind to dissolved metals within the solution that would otherwise inhibit the PCR reaction and hinder the ability to collect proper data for the overall experiment. Heating the cells denatures them and makes them burst, releasing their DNA into solution. The tube is cooled and centrifuged once more. The liquid at the top of the sample after centrifuging is the DNA. (Bottom photo)
PCR Amplification of VMAT2 Gene
Primer was added to the beaded Taq DNA polymerase and buffers. Once the bead was dissolved, DNA was added. The tube was centrifuged, and then added to the PCR machine. The PCR machine cycles through hot and cooler temperatures to denature DNA, enabling primers to bind to DNA, allowing Taq polymerase to add nucleotides, and repeat. This replicated the desired sections of DNA without replicating the entire strand.
Gel Electrophoresis
Dye buffer was added to the PCR tubes to serve as a visual cue for when electrophoresis was to be terminated (the dye moves faster than the DNA so DNA does not run off the gel). A ladder was added into the first well to serve as a template. The ladder has known lengths that the DNA can be compared to. DNA with dye was added into wells 3 through 6.(top left photo) The gel was then run for approximately 30 minutes. After running, the gel was placed under an ultraviolet light and photographed.(bottom right photo) The photograph was used to determine specific lengths of each DNA piece. Samples were cut out of the gel with a fresh razorblade, inserted into a labeled tube, and refrigerated.
Sample Purification
Gel sample purification was done to obtain a clean DNA sample ready for analysis. QG buffer was used to dissolve the non DNA components of the agarose sample. The sample was heated to completely dissolve any leftover agarose not dissolved by QG. Isopropanol was added to the solution to help the DNA bind. The solution was transferred to a 'column' tube to filter the sample. The column was centrifuged to bring the DNA sample onto the white material in the column. The sample is then washed to remove any other trace elements present in the DNA. PE buffer was added to clean the sample of any extra salts that may have been present and would have interfered with sequencing. The last buffer, EB, was used to release the DNA sample from the white column into a clean eppendorf tube for DNA sequencing. The sample was sent to a lab for DNA sequencing.
Statistical Methods
A Chi-square test was used to compare the student class averages to the averaged data from Hamer’s study. This calculation tests the null hypothesis, proving statistical significance between expected and observed results. Our p-value was greater than .05, the cutoff for statistical significance. This means that our data were not significant. There were six categories tested (one for each of the two possible alleles at each of the three pairs of interest). The degrees of freedom were 5 (n-1). Hamer’s data was obtained from http://pharmacogenetics.ucsf.edu/public-results/vmat2/VMAT2-tab.html and the class snp data was simply a total of allele occurrences. Each of these data sets were converted into percentages per base pair and input into the chi-square calculation.
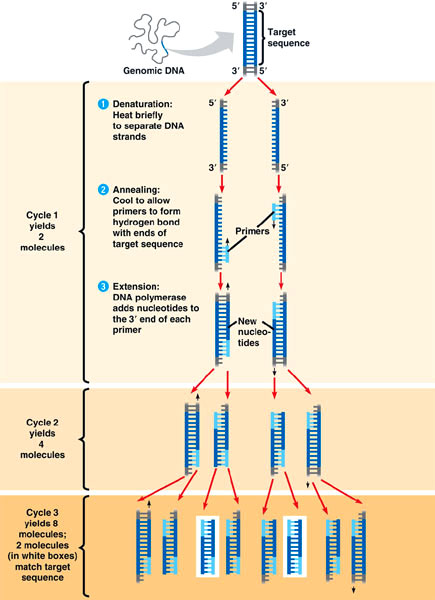
(Figure 1)
PCR Reaction:
The purpose of the PCR reaction is to replicate a specific sequence of DNA without replicating the entire DNA strand. PCR reactions can be carried out using a small amount of DNA. Each cycle of the PCR reaction doubles the amount of DNA.
